
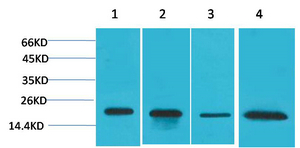
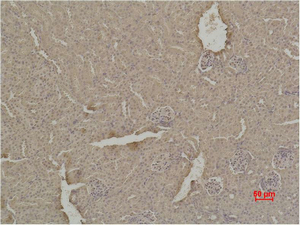
Main InformationTargetSOD1Host SpeciesRabbitReactivityHuman, Mouse, RatApplicationsWB, IHC, IFMW18kD (Observed)Conjugate/ModificationUnmodifiedDetailed InformationRecommended Dilution RatioWB 1:500-1:2000; IHC:1:50-300; IF 1:50-200FormulationPBS, pH 7.4, containing 0.5%BSA, 0.02% sodium azide as Preservative and 50% Glycerol.SpecificityThe antibody detects endogenous SOD1 protein.PurificationThe antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.Storage-15°C to -25°C/1 year(Do not lower than -25°C)MW(Observed)18kDModificationUnmodifiedClonalityPolyclonalIsotypeIgGAntigen&Target InformationImmunogen:Synthesized peptide derived from the Internal region of human SOD-1.Specificity:The antibody detects endogenous SOD1 protein.Gene Name:SOD1Protein Name:Superoxide dismutase [Cu-Zn]Other Name:SOD1 ; Superoxide dismutase [Cu-Zn] ; Superoxide dismutase 1 ; hSod1Background:The protein encoded by this gene binds copper and zinc ions and is one of two isozymes responsible for destroying free superoxide radicals in the body. The encoded isozyme is a soluble cytoplasmic protein, acting as a homodimer to convert naturally-occuring but harmful superoxide radicals to molecular oxygen and hydrogen peroxide. The other isozyme is a mitochondrial protein. Mutations in this gene have been implicated as causes of familial amyotrophic lateral sclerosis. Rare transcript variants have been reported for this gene. [provided by RefSeq, Jul 2008],Function:Catalytic activity:2 superoxide + 2 H(+) = O(2) + H(2)O(2).,cofactor:Binds 1 copper ion per subunit.,cofactor:Binds 1 zinc ion per subunit.,Disease:Defects in SOD1 are the cause of amyotrophic lateral sclerosis type 1 (ALS1) [MIM:105400]. ALS1 is a familial form of amyotrophic lateral sclerosis, a neurodegenerative disorder affecting upper and lower motor neurons and resulting in fatal paralysis. Sensory abnormalities are absent. Death usually occurs within 2 to 5 years. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of cases leading to familial forms.,Function:Destroys radicals which are normally produced within the cells and which are toxic to biological systems.,miscellaneous:The protein (both wild-type and ALS1 variants) has a tendency to form fibrillar aggregates in the absence of the intramolecular disulfide bond or of bound zinc ions. These aggregates may have cytotoxic effects. Zinc binding promotes dimerization and stabilizes the native form.,online information:ALS genetic mutations db,online information:Superoxide dismutase entry,PTM:Unlike wild-type protein, the pathogenics variants ALS1 Arg-38, Arg-47, Arg-86 and Ala-94 are polyubiquitinated by RNF19A; which leads to their proteasomal degradation.,similarity:Belongs to the Cu-Zn superoxide dismutase family.,subunit:Homodimer. The pathogenics variants ALS1 Arg-38, Arg-47, Arg-86 and Ala-94 interact with RNF19A, whereas wild-type protein does not.,Cellular Localization:Cytoplasm . Mitochondrion . Nucleus . Predominantly cytoplasmic; the pathogenic variants ALS1 Arg-86 and Ala-94 gradually aggregates and accumulates in mitochondria. .Tissue Expression:Colon,Fetal brain cortex,Placenta,Research Areas:>>Peroxisome ; >>Longevity regulating pathway - multiple species ; >>Parkinson disease ; >>Amyotrophic lateral sclerosis ; >>Huntington disease ; >>Prion disease ; >>Pathways of neurodegeneration - multiple diseases ; >>Chemical carcinogenesis - reactive oxygen species
商品信息已成功复制,启研竭诚为您服务




